The intricate dance of atoms is pivotal to the very fabric of existence. At the heart of an atom lies a positively charged nucleus—usually comprising protons and neutrons—encircled by a cloud of negatively charged electrons. This configuration not only gives rise to the atom’s identity but also dictates its properties and behaviors in the grander scheme of molecular interactions. When atoms unite to form molecules, they engage in a complex interplay of electron interactions. Understanding these dynamics is crucial for various scientific explorations, including material science and drug development. However, simulating these interactions has historically been one of the most formidable challenges in modern science.
The Computational Challenge: Schrödinger’s Legacy
The roots of this challenge can be traced back to the Schrödinger equation, which serves as a fundamental pillar in quantum mechanics. This equation describes the energy states different configurations of atoms or molecules can inhabit. For chemists and physicists, solving the Schrödinger equation is akin to climbing a steep mountain: it becomes increasingly daunting as the number of atoms in a molecule rises. For any system larger than a few dozen atoms, the computational resources required can spiral out of control—often demanding days of computation, even on cutting-edge supercomputers. When it comes to performing molecular dynamics simulations over extended periods, researchers might find themselves needing to solve the Schrödinger equation thousands of times, an endeavor that becomes impractical.
A Bridge Over Troubled Waters: Machine Learning Interventions
In recent years, researchers have turned to machine learning (ML) as a potential lifeline to ease this computational burden. Rather than directly addressing the Schrödinger equation, ML algorithms aim to predict the outcomes of electron interactions more efficiently. By training on vast quantities of data, ML systems can discern patterns and extrapolate the dynamics of molecular interactions with reduced computational overhead. The underlying struggle for scientists lies in developing algorithms that can teach machines how to anticipate electron behavior without the intricate modeling traditionally required.
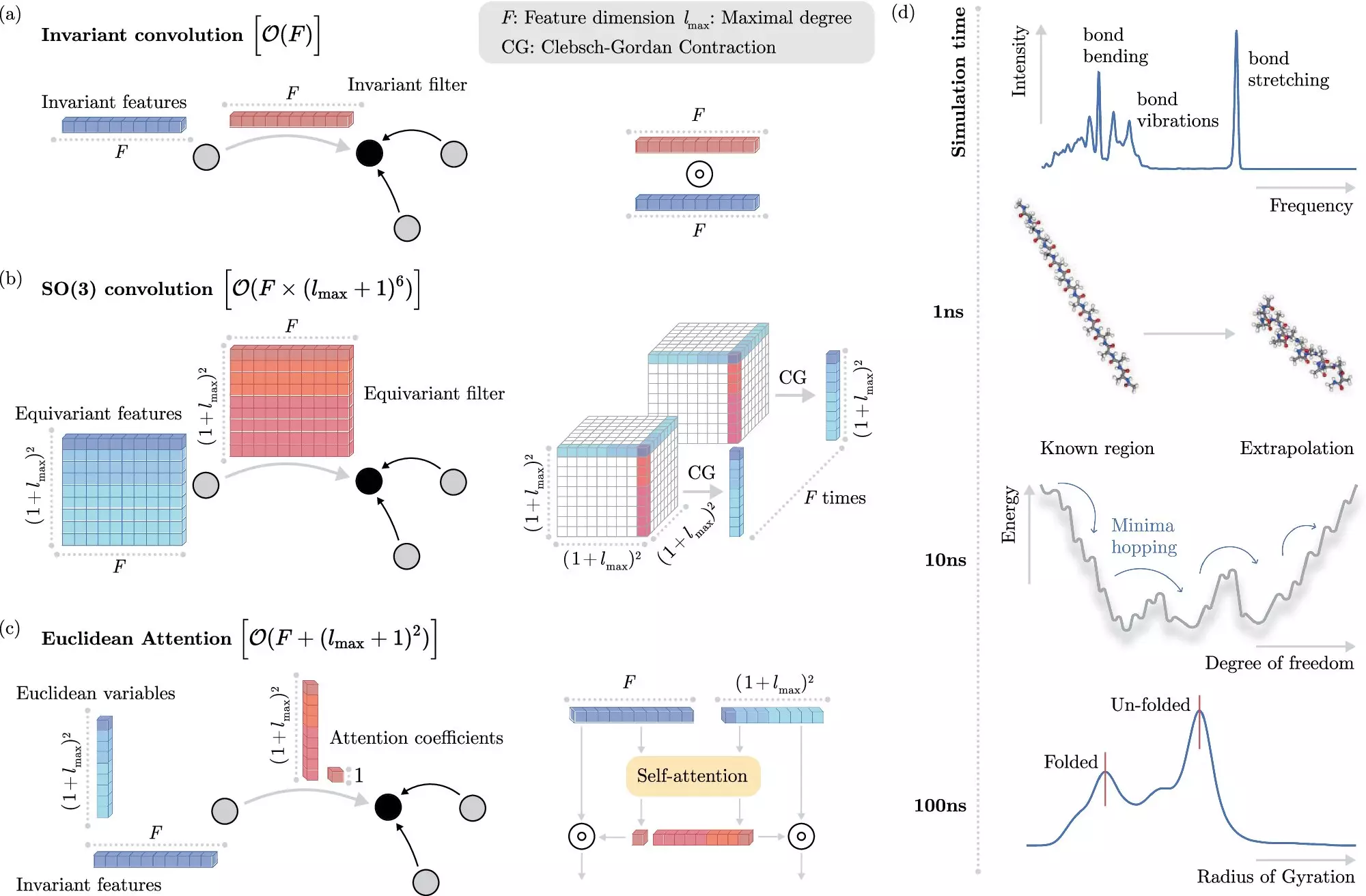
A significant breakthrough in this arena involves recognizing and leveraging the invariances present in molecular systems. In simpler terms, certain molecular properties remain stable, even when the molecules are spatially transformed, provided that the relative positions of the individual atoms are maintained. This realization allows researchers to streamline the learning process, avoiding unnecessary complexities that typically encumber ML models.
Decoupling Complexity: Innovations from BIFOLD and Google DeepMind
Recent advancements from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) and Google DeepMind mark a turning point in this field. The researchers introduced a novel ML algorithm that effectively decouples invariances from the intricate data of a chemical system. This innovation means the algorithm reserves its computational heft for the complexity that truly matters, simplifying the processing of molecular dynamics simulations significantly.
As a result of this leap in efficiency, what once necessitated months of computational power on expansive high-performance clusters can now be executed in mere days on a single computer node. This advancement opens the door for conducting long-time scale simulations that are indispensable for revealing the structure and dynamics of atomic systems. The prospects of meaningful insights into nature’s foundational processes have become tantalizingly close.
Real-World Applications: A Paradigm Shift in Drug Development
One particularly exciting application of this novel algorithm lies in its potential to simulate interactions between molecules and proteins in the human body. By paving the way for precision modeling, researchers may soon be able to pioneer new drug discoveries with minimal physical experimentation. This shift promises not just to save resources—both financial and environmental—but it also positions science to operate with unprecedented efficiency.
For instance, in one of their experiments, the team successfully employed their novel ML approach to analyze docosahexaenoic acid (DHA), a crucial fatty acid integral to human brain structure. Scanning tens of thousands of potential molecular configurations with the traditional quantum mechanical methods would have been a Herculean task, but the ML algorithm made this process feasible and efficient.
Looking Ahead: The Future of Computational Chemistry
As this research progresses, it underscores an essential point: by harmonizing machine learning techniques with established physical principles, scientists can tackle long-standing hurdles within computational chemistry. With future iterations of these algorithms, the focus will shift towards accurately simulating larger, more complex systems, addressing long-range physical interactions that traditional models have struggled with. This evolution promises to not only redefine computational chemistry but also to revolutionize our understanding of molecular dynamics as a whole.
Every step in this direction holds the promise of unveiling the hidden intricacies of nature, unlocking doors to unimaginable discoveries in medicine, materials science, and beyond. The horizon for molecular simulations is expanding, inviting a new era of scientific inquiry bolstered by the synthesis of advanced technology and fundamental physics.
Leave a Reply